
Targets of computational quantum chemistry are expanding to large molecular systems such as functional proteins, molecular assemblies, and complex surface catalyses. However, computing time and resources required for the electronic structure calculations increase rapidly with the size of the molecule. One of the feasible approaches to treat large molecules is the combined quantum mechanics and molecular mechanics (QM/MM) method. In this hybrid approach, the target molecule is divided into QM and MM regions. The electronic structure calculations are performed within the QM region to describe electronic structures and chemical reactions in the ground and excited states. The rest of the molecule is treated by the classical mechanics such as MM method. The interactions between the QM and MM regions are described by using the MM force field (electrostatic, through-bond, and van der Waals interactions). The greatest advantage of the QM/MM approach is the computational efficiency, which realizes the calculations of large scale molecules feasible without losing much accuracy. In our recent study, we developed a QM/MM program and performed geometry optimization for photobiological proteins such as fluorescent proteins[1] and retinal proteins[2].
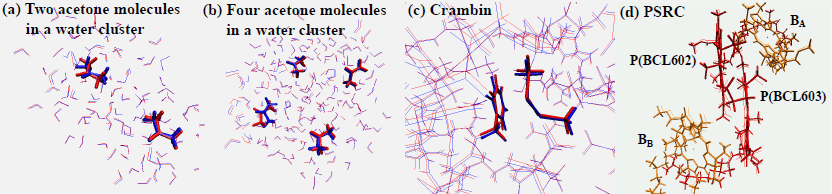
We presented a QM/MM method which can optimize the structure of chromophore aggregate in protein, multi-core (mc) QM/MM method. A QM region is composed of the sum of the QM sub-regions that are small enough to apply practical electronic structure calculations. QM/MM energy gradient calculations are performed for each QM sub-region. Several benchmark examinations were carried out to figure out availabilities and limitations. In the inter-region distances more than 3.5-4.0 angstrom, the mcQM/MM energy gradient is very close to that obtained by the ordinary QM/MM method in which all the QM sub-regions were treated together as a single QM region. In van der Waals complex, the error exponentially drops with the distance, while the error decreases slowly in a hydrogen bonding complex. On the other hand, the optimized structures were reproduced with reasonable accuracy in both cases. The computational efficiency is the best advantage in the mcQM/MM approach, especially when the QM region is significantly large and the QM method used is computationally demanding. With this approach, we could optimize the structures of a bacterial photosynthetic reaction center protein in the ground and excited states, which consists of more than 14000 atoms.
[1]Excited States of GFP Chromophore and Active Site Studied by the SAC-CI method: Effect of Protein-environment and Mutations, J. Hasegawa, K. Fujimoto, B. Swerts, T. Miyahara, and H. Nakatsuji, J. Comp. Chem. 28, 2443-2452 (2007).
[2]Origin of color tuning in human red, green, and blue cone visual pigments: SAC-CI and QM/MM study, K. Fujimoto, J. Hasegawa, and H. Nakatsuji, Chem. Phys. Lett. (2008) in press.
[3]A multi-core QM/MM approach for the geometry optimization of chromophore aggregate in protein, Y. Kiyota, J. Hasegawa, K. Fujimoto, B. Swerts, and H. Nakatsuji, Submitted.